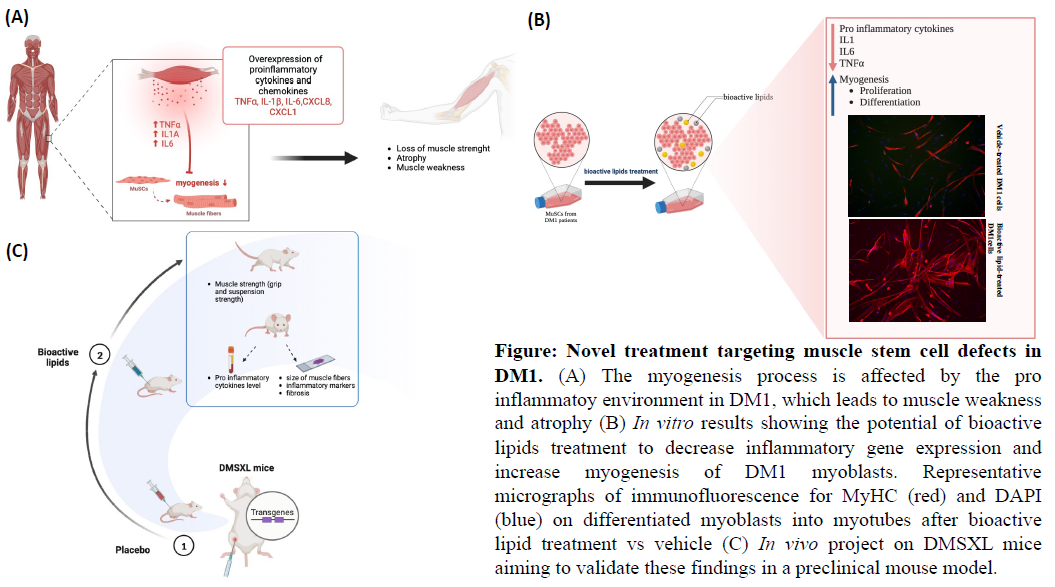
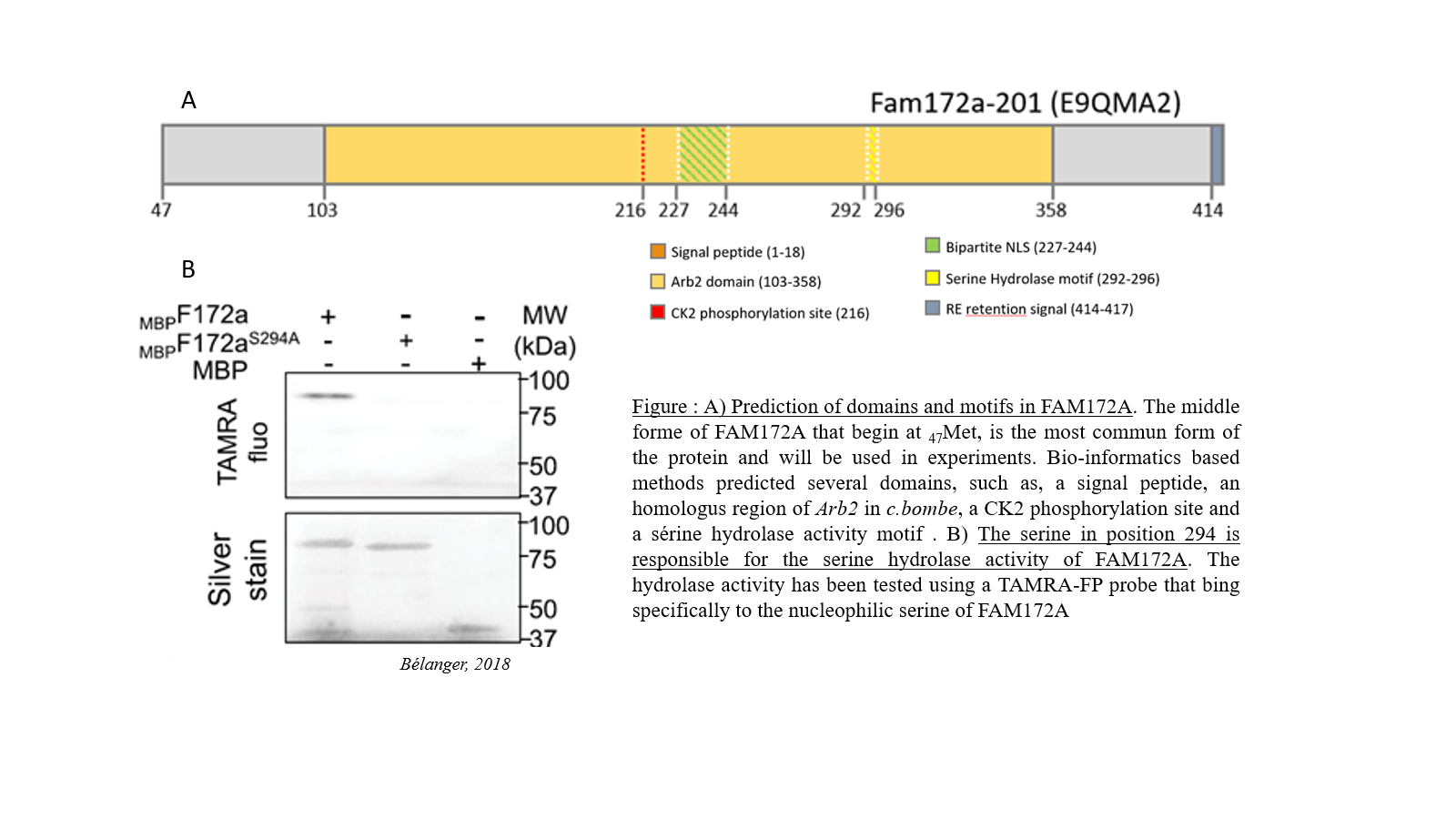
Promote research on orphan diseases
CERMO-FC aim to develop and deepen knowledge about orphan diseases, and to prepare the next generation in this field through education and training.
Thus, the center supports the development of collaborative research projects through grant competitions, and each year promotes the research of a dozen master or doctoral students through scholarships. By supporting its researchers and student members, CERMO-FC wishes to offer them support to obtaining large-scale funding, promoting excellence and success of major scientific breakthroughs in the field of orphan diseases in biomedical and biopharmaceutical research.