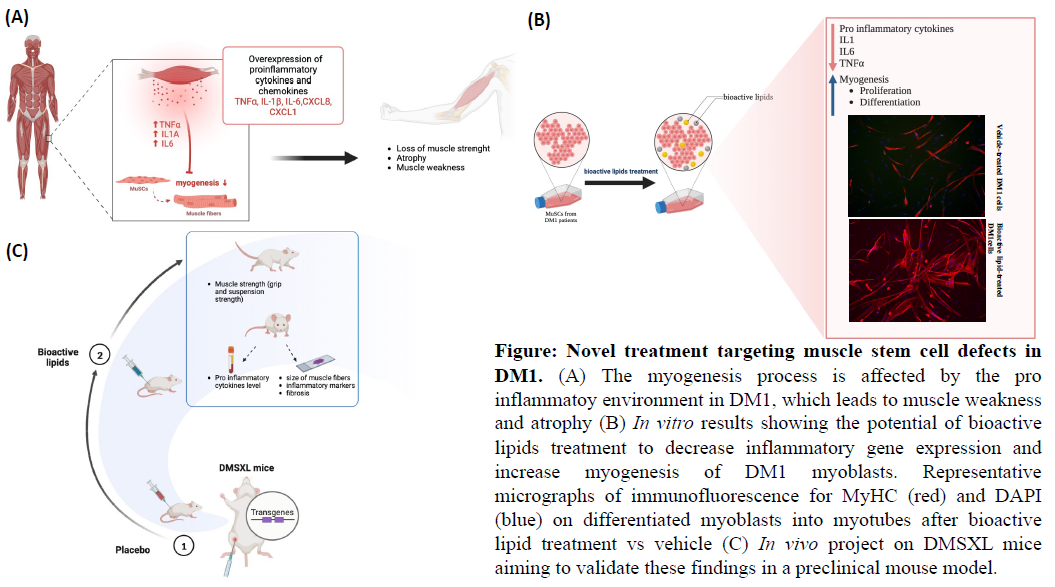
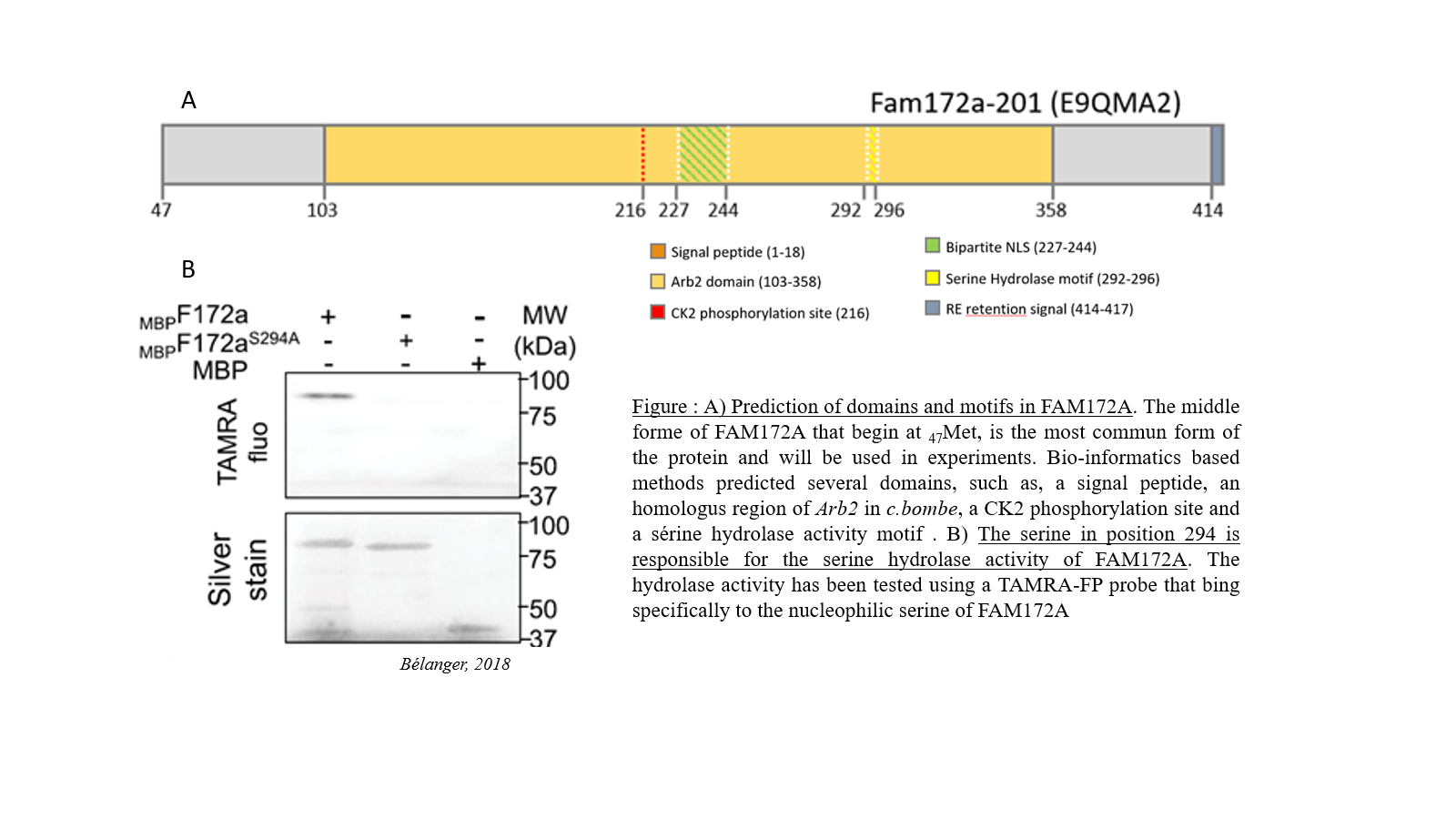
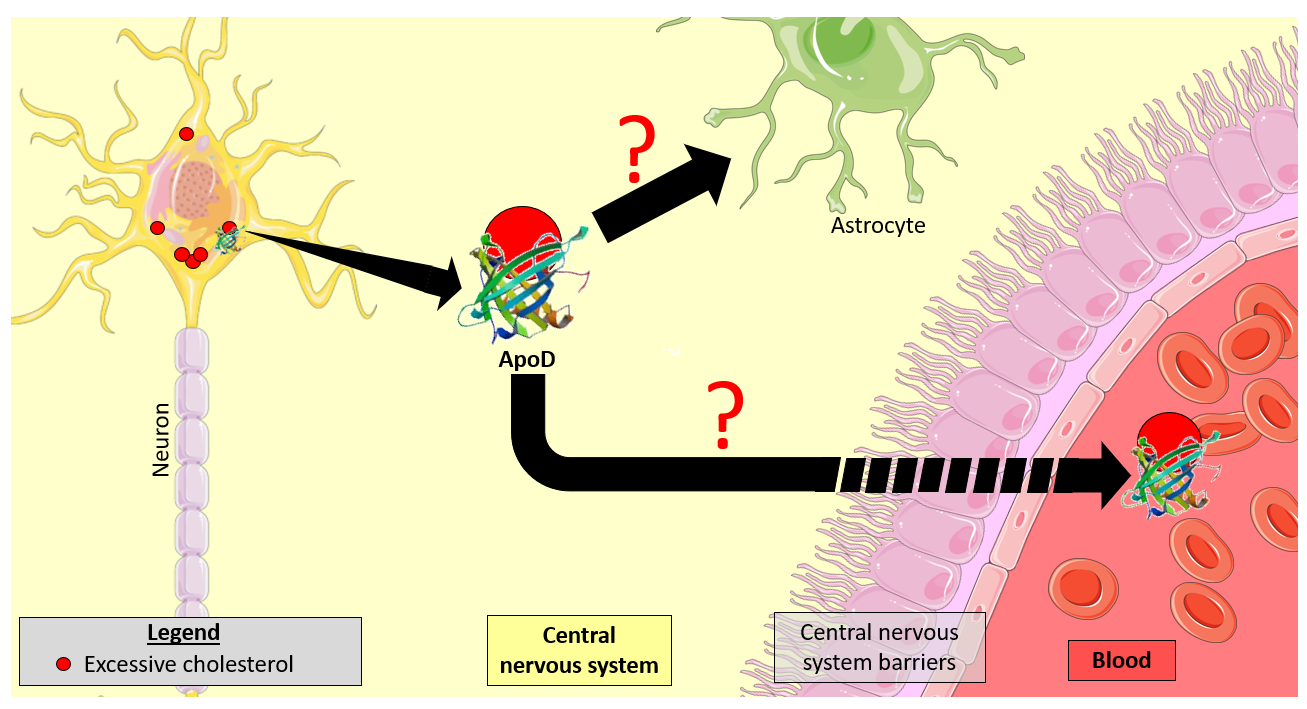
Promouvoir la recherche sur les maladies orphelines
Le CERMO-FC s’est donné comme missions de développer et d’approfondir les connaissances sur les maladies orphelines, et de préparer la relève dans ce domaine par le biais de l’enseignement et de la formation.
Ainsi, le centre soutien le démarrage de projets de recherche en collaboration par le biais de concours de subventions, et valorise chaque année la recherche d’une dizaine d’étudiant-es à la maitrise ou au doctorat en offrant des bourses d’études supérieures. En soutenant ses chercheur-es et étudiant-es membres, le CERMO-FC souhaite leur offrir un tremplin vers l’obtention de financement d’envergure, favorisant l’excellence et la réussite des percées scientifiques majeures dans le domaine des maladies orphelines en recherche biomédicale et biopharmaceutique.